Vivant
- Santé, Médecine et Sciences du Vivant
- Biologie & Biochimie
Sclérose latérale amyotrophique : une piste thérapeutique pour les formes les plus graves
- Tweeter
-
-
1 avis :

{kind=link}
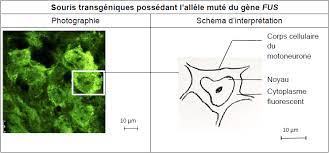
La sclérose latérale amyotrophique (SLA), également nommée maladie de Charcot, est une maladie génétique caractérisée par la dégénérescence des cellules nerveuses qui contrôlent nos muscles : les motoneurones. Chez les malades, ce phénomène se traduit par une paralysie progressive à l’issue fatale. Des mutations du gène FUS sont responsables des cas les plus graves, avec un début précoce (avant l’âge de 40 ans) et une progression rapide de la maladie. Ce gène code pour une protéine de régulation de l’ADN et de l’ARN, habituellement localisée dans le noyau des cellules. Mais les mutations associées à la SLA provoquent une délocalisation de la protéine : on la retrouve alors principalement dans le cytoplasme des cellules, en dehors du noyau. Cette anomalie est à l’origine de la neurodégénérescence.
À l’Université de Strasbourg, Luc Dupuis et son équipe cherchent donc comment réduire la quantité de protéine FUS présente dans le cytoplasme des motoneurones des patients, afin de ralentir la progression de la maladie. Pour bien comprendre leurs travaux, il faut revenir sur les caractéristiques de la protéine FUS elle-même. Sa production est autorégulée : son gène code à la fois pour la protéine et pour des mécanismes qui permettent d’ajuster sa synthèse en fonction de la quantité déjà présente dans le noyau cellulaire.
En situation normale, quand la protéine FUS reste dans le noyau, son taux est stable grâce à ce système : ajoutez plus de vingt copies du gène FUS au génome d’une souris, et le niveau de la protéine n’augmentera que légèrement, sans conséquences pour la santé de l’animal ! En revanche, en cas de mutation, la sortie de la protéine FUS vers le cytoplasme déclenche un signal d’alerte : puisque la concentration de FUS dans le noyau diminue, il faut en produire plus. Mais comme la protéine continue à sortir du noyau, ce mécanisme entraîne une production incessante de FUS, qui s’accumule toujours davantage dans le cytoplasme.
Pour briser ce cercle vicieux, l’équipe de Luc Dupuis propose d’insérer un gène FUS sain et complet dans les motoneurones des malades. Les chercheurs espèrent ainsi augmenter la production de la protéine « normale » dans le noyau, mais aussi activer le système d’autorégulation qui s’appliquerait également à l’expression du gène mutant. Cette approche permettrait alors de réduire la quantité de protéines FUS susceptibles de passer dans le cytoplasme. Les chercheurs ont testé leur stratégie chez des souris porteuses de mutations du gène FUS.
Lorsque les animaux portent une mutation sur chacun des deux allèles du gène, ce qui entraîne leur décès dans les heures suivant leur naissance, l’addition d’un gène FUS sauvage prolonge leur survie jusqu’à l’âge adulte. Lorsque les animaux possèdent un seul allèle du gène FUS muté, comme c’est le cas des patients, ajouter un gène sain supplémentaire prévient l’apparition de la faiblesse musculaire.
Suite à ces premiers résultats encourageants, l’équipe travaille désormais à la mise au point d’une thérapie génique fondée sur ce concept, avec le soutien de la SATT Conectus (société d’accélération du transfert de technologies) à Strasbourg. L’idée est de produire un vecteur viral qui permettra d’apporter un gène FUS thérapeutique aux cellules malades, puis de l’injecter au niveau du cerveau et de la moelle épinière pour qu’il pénètre dans les motoneurones. En premier lieu, cet outil sera développé chez le rongeur. Si les effets escomptés sont au rendez-vous, des essais cliniques pourront être envisagés chez l’Homme.
Article rédigé par Georges Simmonds pour RT Flash
Noter cet article :
Vous serez certainement intéressé par ces articles :

FIV : un nouveau traitement augmente les chances d'implantation de l'embryon
Une étude européenne regroupant des chercheurs espagnols, tchèques et polonais a révélé de bons résultats d’implantation d’embryon avec l’usage d’un médicament non hormonal, qui agit sur l’endomètre,...

Cancer : les survivants ont un risque accru de maladies tout au long de leur vie
Selon une nouvelle étude de l'Université de Linköping (Suède), les personnes ayant survécu à un cancer pendant leur jeunesse courent un risque plus élevé de maladies cardiovasculaires. Elles ont ...

Les flavonols réduisent sensiblement les risques de mortalité
Les flavonols sont un type de flavonoïdes puissants composés bioactifs présents dans presque tous les aliments à base de plantes. Une étude chinoise de l'Université médicale d'Anh montre qu'un ...
Recommander cet article :
- Nombre de consultations : 0
- Publié dans : Biologie & Biochimie
- Partager :