Vivant
- Santé, Médecine et Sciences du Vivant
- Biologie & Biochimie
Publication des premières cartes génétiques des répétitions d’ADN liées à des maladies mortelles
- Tweeter
-
-
1 avis :

{kind=link}
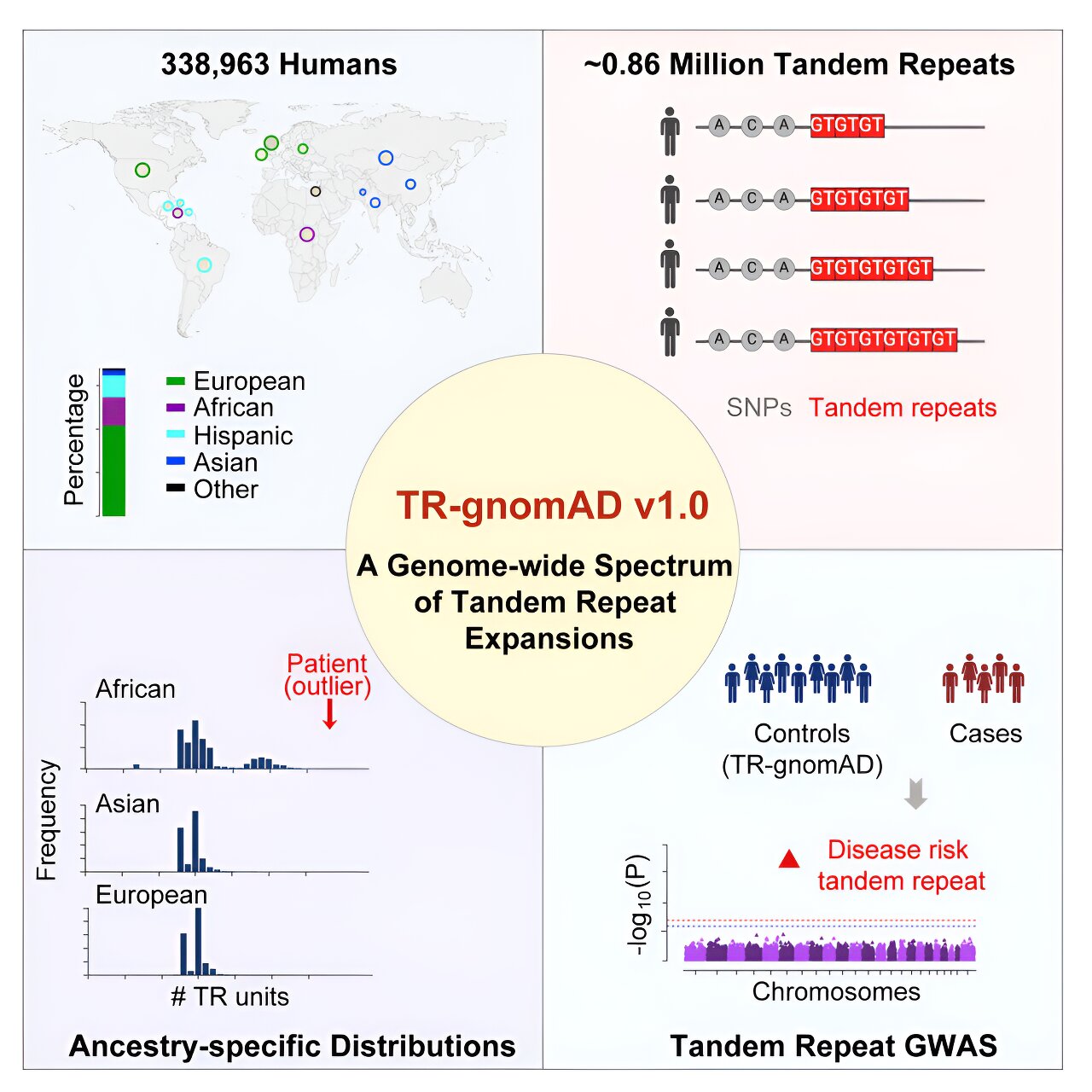
Une équipe de recherche dirigée par l'Université de Californie à Irvine a construit les premières cartes de référence génétique pour de courtes longueurs d'ADN répétées plusieurs fois, connues pour causer plus de 50 maladies humaines mortelles, notamment la sclérose latérale amyotrophique, la maladie de Huntington et des cancers multiples.
La base de données d’agrégation du génome tandem d’UC Irvine permet aux chercheurs d’étudier comment ces mutations – appelées expansions répétées en tandem – sont liées aux maladies, de mieux comprendre les disparités en matière de santé et d’améliorer les diagnostics cliniques. L'étude présente l'UC Irvine TR-gnomAD, qui comble une lacune critique dans les efforts actuels de séquençage du génome des biobanques. Bien que les expansions de TR constituent environ 6 % de notre génome et contribuent de manière substantielle à des conditions congénitales complexes, leur compréhension scientifique reste limitée.
Ce projet révolutionnaire positionne l'UC Irvine comme un leader en génétique humaine et médicale en comblant l'écart critique dans la capacité à interpréter les expansions de TR chez les individus atteints de troubles génétiques. Le TR-gnomAD améliore notre capacité à déterminer comment certaines maladies pourraient affecter divers groupes de personnes en fonction des variations de ces mutations entre les ancêtres. Les sociétés de conseil en génétique peuvent ensuite développer des produits pour interpréter ces informations et signaler avec précision comment certains traits pourraient être liés à différents groupes de personnes et maladies.
Pour créer la base de données, l'équipe a utilisé deux outils logiciels pour analyser les données génomiques de 338 963 participants dans 11 sous-populations. Sur les 0,91 million de TR identifiés, 0,86 million étaient de qualité suffisamment élevée pour être conservés pour une étude plus approfondie. Il a également été découvert que 30,5 pour cent d’entre eux présentaient au moins deux formes alternatives courantes d’un gène provoqué par une mutation située au même endroit sur un chromosome. « Bien que nous ayons réussi à génotyper un nombre important de TR, cela ne représente encore qu'une fraction du nombre total dans le génome humain », a déclaré Li. « Nos prochaines étapes consisteront à donner la priorité à l'intégration d'un plus grand nombre de TR de haute qualité et à inclure davantage d'ascendances sous-représentées, telles que l'Australie, les îles du Pacifique et la Mongolie, à mesure que nous nous rapprochons de la réalisation d'une médecine de précision personnalisée ».
Article rédigé par Georges Simmonds pour RT Flash
Noter cet article :
Vous serez certainement intéressé par ces articles :

Un composé de chardon fait repousser les fibres nerveuses
Des chercheurs de l’Université de Cologne ont identifié un composé du chardon (Cnicus benedictus), la cnicine, qui accélère considérablement la croissance des axones (fibres nerveuses). Ces données, ...

Vers un vaccin thérapeutique à ARN contre les allergies
La société Néovacs développe de nouvelles thérapies pour des maladies inflammatoires et auto-immunes. Elle vient de bénéficier du soutien de l’Agence nationale de la recherche (ANR) à un deuxième ...

Un nouveau modèle de tumeurs cérébrales offre de l'espoir pour les traitements sur le cancer personnalisés
Des scientifiques du Centre de recherche sur le cancer allemand (DKFZ) et de l'Université de Shanghai ont développé une méthode innovante pour la croissance des tumeurs cérébrales de patients ...
Recommander cet article :
- Nombre de consultations : 0
- Publié dans : Biologie & Biochimie
- Partager :